分子シミュレーションによるドラッグディスカバリー
Drug Discoveryは、マテリアルデザインの中で最も発展している領域の一つであり、基礎研究、国家プロジェクトから民間企業プロジェクトまで、その取り組みは多岐に渡ります。
今回は分子シミュレーションに着目したいと思います。
図1:Drug Discoveryの流れ分子シミュレーション
分子シミュレーションとは、分子の動きを再現できるコンピュータシミュレーションの総称です。ですので、実は、ドッキングシミュレーションというのは、分子シミュレーションの一つとなります。図1のように分子シミュレーションと分けているのは、分野としては適切ではありません。ここで指す“分子シミュレーション”は、“ドッキングシミュレーション以外の分子シミュレーション”として捉えて頂ければと思います。
分子シミュレーションは、大きく分けて以下の2つがあります:
■分子動力学(MD)シミュレーション:原子の動きを時々刻々と再現することで、分子の動きを再現できる手法
■ モンテカルロ(MC)シミュレーション:乱数を使ったシミュレーションで、分子の動きの確率分布を得ることができる手法
MCシミュレーションは、分子の静的な性質(結合自由エネルギー、結合構造、etc)を得ることができます。MDシミュレーションは、分子の静的および動的な性質(拡散係数、粘度、etc)も計算することが可能となります。
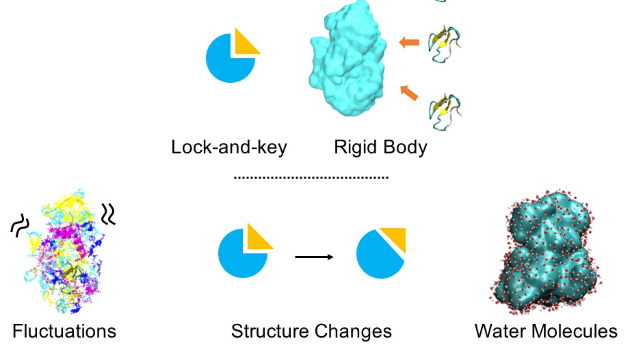
ドッキングシミュレーションとの違い
Drug Discoveryを考える上で、ドッキングシミュレーションと分子シミュレーションの違いを見ていきます。ドッキングシミュレーションは、「タンパク質と薬は構造的にくっつきやすいと良い
0